
Substantial progress continues to be made in the arena of antiretroviral drug development. PRN is again proud to present its annual review of the experimental agents to watch for in the coming months and years. This year’s review is based on a lecture by Dr. Roy M. Gulick, a longtime friend of PRN, and no stranger to the antiretroviral development pipeline.
To date, twenty-two antiretrovirals have been approved by the Food and Drug Administration (FDA) for the treatment of HIV infection. In addition to the development of new drugs and drug classes with unique potency advantages, a number of older antiretrovirals have been reformulated to allow for more simplified dosing. Examples include fosamprenavir (Lexiva) tablets, replacing the amprenavir (Agenerase) liquid capsules; liponavir/ritonavir (Kaletra) tablets, replacing the liquid capsules; and the new tablet formulation of saquinavir (Invirase), replacing the 200-mg capsules of Invirase and the need for Fortovase. Additionally, the development of fixed-dose combination tablets have considerably improved treatment acceptance. For the first time, a widely used complete drug regimen is available to take as one pill once per day. Atripla, a fixed-dose combination drug containing daily doses of tenofovir (Viread), emtricitabine (Emtriva), and efavirenz (Sustiva), was approved by the FDA on July 12, 2006.
Even with increasingly simplified treatment regimens, challenges still remain in finding products with minimal toxicity and optimized resistance profiles. To achieve optimal viral suppression, there is also a need for agents that penetrate viral reservoirs and target new portions of the HIV lifecycle. Fortunately, the antiretroviral drug pipeline contains several promising agents that may address these needs.
A summary of these agents is shown in Table 1. Some of these are new members of existing drug classes, including reverse transcriptase inhibitors and protease inhibitors, and some are members of new drug classes, such as integrase inhibitors.
| NUCLEOSIDE REVERSE TRANSCRIPTASE INHIBITORS (NRTI) | Top of page |
Dr. Gulick described two NRTIs in development: dexelvucitabine, formerly Reverset and developed by Incyte, and apricitabine, being developed by Shire Pharmaceuticals. There is, unfortunately, little need to review the development of dexelvucitabine. On April 3, 2006, Incyte announced that it had discontinued all clinical trials of the drug, due to a high frequency (>15%) of grade 4 hyperlipasemia, a marker of pancreatic inflammation (Incyte, 2006).
Apricitabine
Apricitabine is a cytosine analogue that, in vitro, is active against both wild-type virus and HIV with reverse transcriptase harboring the M184V mutation. However, it is not active against HIV containing the RT mutations K65R, V75I, and Q151.
Apricitabine has a bioavailability of 85% to 90%, with linear pharmacokinetics and a long extracellular half-life of approximately 10 hours (Zhu, 2003). During apricitabine’s development, a serious drug interaction with lamivudine (Epivir) was noted. Although the plasma concentrations of apricitabine were unaffected by coadministration of lamivudine, the intracellular concentrations of apricitabine were reduced by approximately sixfold. Additionally, the 50% inhibitory concentration (IC50) of apricitabine against HIV with the M184V mutation was increased 2- to 4-fold in the presence of lamivudine (Bethell, 2004). Taken together, these data suggest that coadministration of apricitabine with lamivudine will be an unlikely option.
Dr. Gulick reviewed a phase I placebo-controlled, dose-ranging study involving 62 HIV-positive, treatment-naive volunteers with HIV-RNA levels between 5,000 and 100,000 copies/mL and CD4+ counts above 250 cells/mm3. The patients were randomized to six doses of apricitabine given once or twice daily, or a matched placebo. Dose-dependent viral load reductions of up to 1.6 log10 copies/mL were seen at the end of 10 days. Apricitabine was well tolerated with no development of resistance mutations over the short 10-day study (Cahn, 2006). Phase II studies are currently under way.
| NON-NUCLEOSIDE REVERSE TRANSCRIPTASE INHIBITORS (NNRTI) | Top of page |
Among several investigational agents, two NNRTIs are currently being developed by Tibotec Therapeutics for drug-resistant viruses: etravirine (TMC-125) and rilpivirine (TMC-278).
Etravirine (TMC-125)
Etravirine is a diaminopyrimidine (DAPY) compound. The DAPY structure gives the molecule flexibility in binding to the NNRTI-binding site of reverse transcriptase, which theoretically allows it to be active in the presence of mutations conferring resistance to currently available NNRTIs.
In vitro, etravirine demonstrates potent activity against wild-type HIV; it also has some demonstrated activity against HIV-2 (in contrast to the currently approved NNRTIs). Most importantly, it has activity against many NNRTI-resistant viral strains. Like other NNRTIs, etravirine is metabolized by the hepatic cytochrome P450 CYP3A4 enzyme system and, therefore, will likely have clinical challenges due to drug interactions. Lopinavir/ritonavir decreases the concentrations of etravirine by 30%, and tipranavir/ritonavir decreases concentrations by more than 75% (Schöller, 2006). However, the recently FDA-approved darunavir (Prezista) has no effect on etravirine exposures, so these two compounds could potentially be used together. An ongoing study is exploring the clinical utility of this combination.
In a seven-day, placebo-controlled study of 19 treatment-naive HIV-infected patients, a reduction of 1.9 log10 copies/mL in HIV-RNA was seen (Gruzdev, 2003). In an eight-day study of sixteen treatment-experienced patients who had previously failed an NNRTI-based regimen (19% efavirenz, 81% nevirapine), a reduction of greater than 1.0 log10copies/mL in HIV-RNA was seen.
Most recently, a Phase II study (Study TMC125-C223) evaluated 199 NNRTI-experienced patients who had documented NNRTI resistance and more than three primary protease mutations. The mean baseline viral load was 48,000 copies/mL, the CD4+ count was approximately 100 cells/mm3,and median baseline phenotypic susceptibility was decreased 41-fold for efavirenz, 68-fold for nevirapine, and 1.6-fold for etravirine. Patients were randomized 1:2:2 to receive active control (the best regimen from currently approved antiretroviral agents), an etravirine 400-mg, BID-based regimen, or an etravirine 800-mg, BID-based regimen. In the etravirine arms, the clinician selected an optimized nucleoside regimen, and could also use lopinavir/ritonavir in addition to enfuvirtide (Grossman, 2005). After 24 weeks of therapy, the control group achieved a reduction of 0.2 log10 copies/mL in HIV-RNA, while the etravirine 400- and 800-mg BID groups achieved HIV-RNA reductions of 1.0 and 1.2 log10 copies/mL, respectively. A greater number of NNRTI mutations was associated with a decreased virologic response.
According to a subset analysis of 79 patients receiving etravirine at 800 mg BID from this study, reported by Dr. Johan Vingerhoets of Tibotec at the 15th International HIV Drug Resistance Workshop, held in June in Sitges, Spain, patients with HIV containing the K103N mutation treated with etravirine plus optimized background therapy (OBT) experienced a viral load reduction of 1.43 log10 copies/mL after 24 weeks of therapy, compared with an HIV-RNA reduction of 1.40 log10 copies/mL in patients without K103N (Vingerhoets, 2006). Among patients with HIV containing the Y181C mutation, there was a decrease of 0.86 log10 copies/mL after 24 weeks of treatment, compared with a viral load decrease of 1.70 log10 copies/mL in the patients without Y181C. Since Y181C is usually associated with previous nevirapine (Viramune) use, and rarely associated with previous efavirenz (Sustiva) use, these data suggest that, while patients who have failed either of these currently available NNRTIs may benefit from etravirine therapy, those who only have the K103N mutation may fare better. Etravirine is currently in Phase III development and is now available in an expanded access program.
Rilpivirine (TMC-278)
Rilpivirine (formerly TMC-278) is also a flexible DAPY compound that has demonstrated in vitro activity against 3500 NNRTI-resistant clinical isolates (de Bethune, 2005). It is well absorbed with a half-life of 38 hours, which is more than adequate for once-daily dosing. In a phase IIa study, 47 treatment-naive patients were randomized to receive doses of 25, 50,100, and 150 mg of rilpivirine or placebo daily for seven days. All of the doses tested had similar antiviral activity, and reduced HIV-RNA by approximately 1.2 log10 copies/mL (Goebel, 2005). This compound is continuing in phase II clinical development.
| PROTEASE INHIBITORS | Top of page |
Two new protease inhibitors (PIs) are currently of interest: darunavir, manufactured by Tibotec, and brecanavir, being developed by GlaxoSmithKline.
{kind=link}
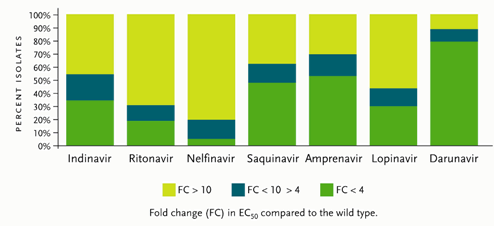
Figure 1.Antiviral Activity of Different Protease Inhibitors Tested Against a Panel of 1,501 Recent Recombinant Clinical Isolates Resistant to At Least One Protease Inhibitor.
Reprinted with permission from De Meyer S, et al. Antimicrob Agents Chemother 2005; 49:2314–2321.
Darunavir (Prezista)
On June 23, 2006, the FDA granted Tibotec accelerated approval of darunavir for treatment-experienced adults who are not responding to treatment with other antiretroviral drugs. Darunavir has been classified as a “resistant-repellant” protease inhibitor, since it has been difficult to develop darunavir resistance after multiple passages of the virus in vitro. Studied with over 5000 isolates from patients failing PIs (some with three or more primary PI mutations), darunavir showed activity against more than 50% of these viruses, with a less-than-4–fold change in 50% effective concentration (EC50) (see Figure 1). Darunavir is currently approved at a dose of 600 mg taken twice daily in combination with ritonavir (Norvir) 100 mg dosed twice daily. Since darunavir is metabolized by the hepatic CYP3A enzyme system, ritonavir enhances the blood concentrations of darunavir. It should be taken with food, although the quantity and type of food was not specified.
The approval of darunavir was based on the results of two randomized studies called POWER I (Study TMC114-C213) and POWER II (TMC114-C202) and a third single-arm study, POWER III. The randomized studies were of similar design and conducted in triple-class-experienced patients with at least one primary PI mutation and study entry viral loads of at least 1000 copies/mL. POWER II, however, enrolled subjects with more advanced HIV disease (Wilkin, 2005).
Participants in both POWER I and II studies received an optimized background therapy that included NRTIs with or without enfuvirtide and were then randomized to either a control arm, consisting of an investigator-selected ritonavir-boosted PI regimen, or one of four darunavir/ritonavir arms: 400/100 or 800/100 mg once daily, or 400/100 or 600/100 mg twice daily.
The combined 48-week data from POWER I and II were reported at the XVI International AIDS Conference (IAC) in Toronto, Canada (Lazzarin, 2006). The reported data reflected 110 patients who had reached 48 weeks of treatment in the darunavir/ritonavir groups, and 120 patients who had reached 48 weeks of treatment in the comparator PI/ritonavir groups.
Sixty-one percent of patients taking a darunavir-based regimen had viral loads that were at least 1 log10 copies/mL below baseline. In the comparator PI groups, 15% had a similar viral load response after 48 weeks of treatment. As for undetectable viral loads, 46% of the darunavir-treated patients had HIV-RNA levels below 50 copies/mL after 48 weeks, compared with 10% of the comparator PI/ritonavir-treated patients.
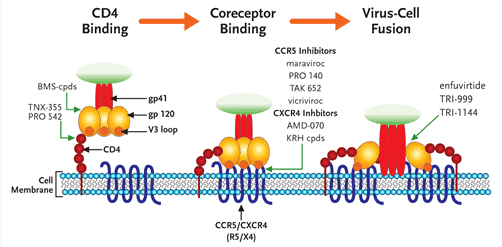
FIGURE 2. A Schematic Illustration of HIV Entry and the Steps Prevented by Different Entry Inhibitors
HIV entry can be divided into 3 discrete steps: attachment of the viral glycoprotein (gp) 120 to the CD4 receptor, followed by subtle conformational changes in gp120 which expose structural elements on the V3 loop that bind to coreceptor, either CCR5 or CXCR4. This induces a structural rearrangement in gp41 which inserts a hydrophobic fusion peptide region into the target cell membrane. This brings the virus and cell membrane in close apposition to initiate fusion and ultimately entry of the virus into the target cell. HIV Entry Inhibitors can be thought of as 3 distinct subclasses, each targeting one of the 3 previous steps. Small molecules that are furthest along in development are highlighted.
Modified with permission from an original image by Robert W Doms, MD, PHD.
Encouraging CD4+ cell count data were reported, as well. After 48 weeks of treatment, darunavir/ritonavir-treated patients experienced a median increase of 102 CD4+ cells/mm3, compared with an increase of approximately 19 CD4+ cells /mm3 in the comparator PI/ritonavir groups.
Thus far, the most common adverse effects in the Prezista-treated patients, compared with those in the control group include, respectively, diarrhea (20% vs 28%), nausea (18% vs 13%), headache (15% vs 20%), and fatigue (12% vs 17%). While hyperlipidemia has been noted in darunavir/ritonavir-treated patients in POWER I II, the increases did not appear to be any more common when compared with those receiving a comparator PI/ritonavir-based regimen.
The POWER investigators plan to publish final data from POWER 1 and II based on 96-week follow-up results. However, the study will continue to follow patients for a total of 144 weeks.
Brecanavir
Brecanavir is an investigational PI that also has activity against viruses resistant to currently available PIs. While it has poor (<5%) bioavailability alone, ritonavir enhances brecanavir concentrations six- to 30-fold. In vitro, brecanavir exhibited activity against 55 viral isolates from PI- experienced patients (Reddy, 2003).
Twenty-four week data from an ongoing 48-week phase II pilot clinical trial of brecanavir are available (Ward, 2005). Thirty-one patients—25 of whom were naive to antiretroviral therapy and six of whom had previously failed a PI-based regimen—received brecanavir/ritonavir 300 mg/100 mg BID combined with two NRTIs. At week 24, 77% of participants had HIV-RNA levels below 50 copies/mL. The 25 treatment-naive patients had an average viral load reduction of 3.3 log10 copies/mL, while the six patients with PI resistance had an average viral load reduction of 2.2 log10 copies/mL. CD4+ counts increased by an average of 84 cells/mm3 among all participants. No serious adverse effects have been reported, although median increases in total cholesterol of 31 mg/dL and in triglycerides of 62 mg/dL were seen. Additional studies of brecanavir are anticipated.
| ENTRY INHIBITORS | Top of page |
The process of viral entry to the host cell can be separated into three substeps (see Figure 2). The virus first binds to CD4+ cells through an interaction between the viral membrane glycoprotein 120 (gp120) and the CD4 receptor on the surface of the cell. This binding induces a conformational change in gp120 that allows binding to a second cellular receptor, the coreceptor or chemokine receptor (which binds either the chemokines CCR5 or CXCR4, or both). Finally, the virus membrane fuses to the cell membrane. The only currently approved agent that blocks this process is enfurvitide, aninhibitor of fusion. While there are several compounds in development to inhibit the first stage of this process, CD4 receptor attachment, the compounds farthest along in clinical development are the coreceptor (chemokine receptor)inhibitors maraviroc and vicriviroc.
For these compounds, it is important to take into consideration the tropism of the virus in patients. Most patients become infected with R5-tropic virus, which specifically binds to the CCR5 coreceptor. Over time, some patients may develop X4-tropic virus, which binds to the CXCR4 coreceptor. In various trials and cohorts, over 80% of treatment-naive patients have shown R5-only tropic virus, 0.1% have X4-only tropic virus, and theremainder have a mixed R5/X4-tropic virus population (Demarest, 2004; Brumme,2005; Moyle 2005). In treatment-experienced patients, however, 50% to 75% have R5-only virus, 2% to 5% have X4-only virus, and the remainder have a mixed R5/X4 virus population (Demerest, 2004; Moyle, 2005; Melby, 2005; Wilkin 2006).
Maraviroc
Maraviroc, being developed by Pfizer, is currently in phase III clinical trials. This compound has demonstrated activity against multidrug-resistant virus, and the suggestion of activity against HIV resistant to other CCR5 inhibitors. Maraviroc is metabolized by the hepatic CYP3A enzyme system and has associated drug interactions: ritonavir enhances maraviroc drug exposure, efavirenz reduces maraviroc exposure, and nevirapine increases maraviroc exposure (Muirhead, 2005).
In a ten-day, phase I study, 82 patients with R5-tropic virus were randomized to one of a number of doses of maraviroc. Patients given the highest dose, 800 mg twice daily, had a reduction of over 1.5 log10 copies/mL in viral load that was durable for up to days after the discontinuation of the drug (Fätkenheuer, 2005).
A current phase IIb/III dose-ranging study in treatment-naive patients is comparing the efficacy of maraviroc (300 mg BID) with efavirenz, both of which are being given in combination with zidovudine/lamivudine (Combivir). Recently, in this IIb/III trial, Pfizer reported one case of severe hepatotoxicity requiring a liver transplant. However, this patient was taking a number of other potentially hepatotoxic medications (including isoniazid), and the noted hepatotoxicity could not be definitively linked to the use of maraviroc (Mayer, 2005). An extensive investigation of more than 1500 patients who have taken maraviroc failed to find any other clear cases of maraviroc-associated hepatotoxicity. Based on these data, the data safety monitoring board recommended that the three phase IIb/III clinical studies in treatment-naive and -experienced patients continue as currently designed. An expanded-access program is planned.
Vicriviroc
Schering-Plough’s vicriviroc is a CCR5 inhibitor in a somewhat earlier stage of development. It is more than 89% bioavailable and has a 27-hour half-life, meaning that once-daily oral dosing is likely. It is also metabolized through the hepatic CYP3A enzyme system, and has drug interaction potential. For example, ritonavir increases vicriviroc concentrations two to five times, while efavirenz and nevirapine decrease its concentrations by 70% to 80% (Strizki, 2005).
A phase I, 14-day study of vicriviroc randomized 48 patients who had not taken antiretroviral drugs for at least eight weeks and who had CD4+ counts of at least 200/mm3 to receive vicriviroc at 10, 25, or 50 mg twice daily or matching placebo. The study demonstrated reductions of up to 1.6 log10 copies/ml HIV RNA at the highest vicriviroc dose, compared with no change in the placebo group (Schurmann, 2004).
A phase II international study evaluated three doses of vicriviroc monotherapy (25, 50, or 75 mg once daily) versus placebo for 14 days in treatment-naive patients, followed by the addition of zidovudine/lamivudine for the vicriviroc-treated arms, and zidovudine/lamivudine plus efavirenz for the placebo group. Participants were treatment naive and R5 tropic. Viral load reductions at day 14 of the monotherapy phase were 0.9 log10, 1.2 log10, and 1.3 log10 copies/mL for the vicriviroc doses of 25, 50, and 75 mg, respectively (Greaves, 2006). The percentage of patients with viral load rebound during the follow-up period also appeared to be dose-dependent: 56% for the 25-mg dose,41% for the 50-mg dose, and 17% for the 75-mg dose. These were substantially different from the 4% rebound in the efavirenz-based control arm. Based on these data, the study of vicriviroc in treatment-naive patients has been suspended. However, studies in treatment-experienced patients are continuing as scheduled.
In ACTG 5211, investigators randomized 118 treatment-experienced patients who were failing their current ritonavir-containing regimen to add vicriviroc at 5, 10, or 15 mg daily or matching placebo for 14 days and then optimize their background antiretroviral regimen based on treatment history and drug resistance testing (Gulick, 2006). The vicriviroc 5-mg arm was stopped early due to trends towards suboptimal antiretroviral activity and more coreceptor switching. At 24 weeks, the vicriviroc 10- and 15-mg arms were associated with a reduction of 1.7 to 1.9 log10 copies/ml HIV RNA, compared with a reduction of 0.3 log10 copies in the placebo group. A total of five patients randomized to vicriviroc developed malignancies (2 Hodgkin’s lymphomas, two non-Hodgkin’s lymphomas and one gastric carcinoma), but causality was uncertain in this population with advanced HIV disease.
Also of note, there has been some evidence of a switch from R5- to X4-tropic virus, or to a dual/mixed tropism viral population among participants in the early clinical trials of these compounds. The clinical significance of this switch is unclear, but there is concern that the emergence of X4 virus could be associated with rapid progression of HIV infection.
In a phase II study reported at the IAC in Toronto, there was a significant switch to X4 virus in maraviroc-treated patients who entered the trial with dual/mixed tropic virus (Mayer, 2006). While this switch translated into limited antiviral activity in patients receiving a maraviroc-based regimen compared with those who were in a control group, CD4+ cell counts were actually higher by the end of the study in the maraviroc treatment groups, suggesting that tropism switch may not be associated with more rapid disease progression.
Whether or not tropism switch is harmful, it has raised interest in combining CCR5 and CXCR4 inhibitors in future antiretroviral drug regimens to prevent and suppress any potential shift from R5 to X4 virus.
To this end, there is currently one CXCR4 inhibitor in clinical development; AMD 070, being developed by AnorMED. The compound has shown in vitro synergy with AMD 887, an investigational CCR5 inhibitor (Schols, 2004). A phase I dose-ranging, safety study conducted in 12 healthy volunteers has demonstrated that a single dose (50-400 mg) of AMD 070 was well tolerated (Stone, 2004). A subsequent phase IIa trial was initiated in March of 2005 with the intention of enrolling up to 48 patients who are HIV positive. The unpublished data released in February of 2006 and involving eight R5 and X4 dual-tropic volunteers, showed a reduction of 1.3 log10 copies/mL in X4 viral load in four participants after 10 days of monotherapy (AnorMED, 2006). Ongoing trials will evaluate long-term efficacy of AMD 070 in patients with a dual-tropic viral population.
| INTEGRASE INHIBITORS | Top of page |
Integration of viral DNA into the host genome is a complex, 3-step process (see Figure 3). The HIV integrase removes two deoxynucleotides at the end of the viral DNA strand and subsequently ligates it to the host chromosomal DNA. The viral-specific integrase enzyme can be targeted much like HIV reverse transcriptase or HIV protease. The group of integrase inhibitors currently in clinical development (MK-0518 and GS-9137) interferes specifically with the last step of the integration process: strand transfer.
MK-0518
Merck’s MK-0518 is primarily metabolized by glucuronidation, meaning that drug interactions are expected to be minimal. In phase I 10-day, monotherapy studies involving treatment-naive patients, MK-0518 resulted in a reduction of 1.7 to 2.2 log10 copies/mL in HIV-RNA (Morales-Ramirez, 2005).
In a subsequent phase II study, 167 heavily treatment-experienced patients were randomized to an optimized background regimen plus either placebo or one of three daily doses of MK-0518: 200, 400, or 600 mg (Grinsztejn, 2006). In preliminary results at 16 weeks, the placebo group had an average viral load reduction of 0.5 log10 copies/mL, while the groups that added the integrase inhibitor had a reduction of greater than 2 log10 copies/mL in viral load. Additionally, nearly 60% of patients on the three MK-0518 arms of the trial achieved viral load reduction to less than 50 copies/mL at week 16.
Twenty-four–week data from a 48-week, phase II study were reported at the XVI IAC by Dr. Martin Markowitz of the Aaron Diamond AIDS Research Center (ADARC) (Markowitz, 2006). The study enrolled 198 treatment-naive patients. The patients were randomized to one of four MK-0518 doses—the same doses explored in the phase I study—or efavirenz. All patients also received tenofovir (Viread) and lamivudine.
After 24 weeks of therapy, 85% to 95% of patients receiving an MK-0518–based regimen had viral loads below 50 copies/mL, compared with 92% in the efavirenz treatment group. CD4+ counts, ranging from 271 to 314 cells/mm3 at baseline, increased in all patients after 24 weeks of treatment. Among patients in the MK-0518 groups, CD4+ counts increased by 139 to 175 cells/mm3. In the efavirenz group, CD4+ counts increased by 112 cells/mm3. Neither the HIV-RNA differences nor the CD4+ cell count differences between the MK-0518 treatment groups and the efavirenz treatment group were statistically significant.
Thus far, treatment with MK-0518 or efavirenz seems to be well tolerated. Nausea, dizziness, and headache appear to be the most frequently reported adverse effects. The only possible treatment-related toxicity of concern was in a patient in the 600-mg MK-0518 group who discontinued therapy due to substantially increased liver enzymes.
Phase III trials are currently enrolling, and an Expanded-Access Program (EAP) has been available since September of 2006. More information on the EAP can be obtained at www.earmrk.com.
GS-9137
GS-9137, being developed by Gilead, also reported early clinical results. This compound is metabolized by the hepatic CYP3A enzyme system. Since ritonavir increases GS-9137 exposure 20-fold, these two agents are being developed together. A phase I study, involving 40 patients who were either treatment naive or treatment experienced, randomized subjects to receive one of four doses of GS-9137 monotherapy, one dose of ritonavir-boosted monotherapy, or placebo for 10 days (DeJesus, 2006). The GS-9137/ritonavir 50-/100-mg, once-daily dose performed best with a reduction of greater than 2 log10 copies/mL in HIV-RNA at day 11, and a substantial antiviral effect lasting between seven and 14 days following treatment discontinuation. This compound is currently in phase II clinical trials.
| MATURATION/ASSEMBLY INHIBITORS | Top of page |
The final step needed for HIV viral assembly is the cleaving and processing of the long precursor gag protein. This takes place after viral budding from infected CD4+ cells. The very last cut cleaves the p25 capsid precursor protein, creating the p24 capsid protein, which is needed for the full maturation of the virus. Without the assembly of the viral capsid, the HIV virion is not infectious and will be cleared from the body. Bevirimat is the first maturation inhibitor currently under development.
Bevirimat
Bevirimat, from Panacos, is currently in phase IIb clinical development. This compound is primarily metabolized by glucuronidation. Its half-life of approximately 60 to 70 hours easily supports once-daily dosing. Bevirimat appears to have a high genetic barrier to resistance, with 10 to 25 serial passages required for the emergence of resistance in vitro (Salzwedel, 2004). No clinical resistance has been seen thus far in phase II trial participants receiving monotherapy (Adamson, 2006).
In the phase IIa trial, 32 treatment-experienced patients were randomized to receive one of four doses of bevirimat or placebo for 10 days. The group receiving the highest dose, 200 mg once-daily, achieved the best viral load reduction of 1.1 log10 copies/mL at day 11 (Smith, 2006). Adverse events have been limited to mild to moderate gastrointestinal discomfort, including diarrhea, loose stools, and nausea. No laboratory abnormalities have been seen thus far.
The FDA has requested that higher doses be explored, and a 14-day, phase IIb, dose-escalating study will randomize treatment-experienced patients to one of five doses (starting at 400 mg once-daily). Responders will then have an optimized background regimen added and be followed for 10 weeks. Phase III trial enrollment is expected to follow.
| Conclusion | Top of page |
Several new antiretroviral agents are under development. These agents either expand benefits to existing drug classes, such as the reverse transcriptase inhibitors and PIs, or are in novel drug classes, such as integrase inhibitors and maturation inhibitors. To summarize, Dr. Gulick put the current situation into perspective: “The pipeline for investigational agents for HIV is full.”
| References | Top of page |